Last Updated on March 20, 2026
Curtis Haavi
Molecular Biologist & Geneticist
16th March 2026
Abstract
Traditional bulk transcriptomic methods average gene expression across heterogeneous cell populations, masking rare subtypes and transient states that drive complex pathologies. Single-cell RNA sequencing (scRNA-seq) overcomes this limitation by capturing gene expression at cellular resolution through molecular barcoding. Strategic platform selection is essential, as technical variation in capture chemistry and transcript detection can significantly influence biological conclusions. As scRNA-seq transitions from academic prototyping to a diverse commercial landscape, systematic evaluations are critical to guide the selection of technologies tailored to specific research objectives. Comparative benchmarking of leading commercial systems shows that droplet- and microwell-based platforms maximize molecular sensitivity, while combinatorial indexing delivers unparalleled throughput. These differences are exemplified through high-resolution mapping of somatic evolution in gynecological malignancies and the immunopathology of chronic inflammatory disorders and renal disease. Integrating these cellular maps with emerging spatial and multi-omic modalities represents a paradigm shift toward fully personalized precision medicine.
Introduction
For decades, molecular biology and clinical genomics have relied on bulk tissue measurements that average signals across millions of cells. Although these approaches have yielded foundational insights into gene regulation and disease mechanisms, they obscure the cellular heterogeneity that underlies development, immunity, and pathology. Diseases traditionally classified by organ system or histology are increasingly recognized as complex ecosystems composed of dynamic and interacting cellular states.
The emergence of single-cell RNA sequencing (scRNA-seq) represents a technological and conceptual inflection point. By resolving transcriptional programs at cellular resolution, scRNA-seq has revealed previously unrecognized cell populations, transient intermediate states, and rare pathogenic subclones that drive disease progression and therapeutic resistance. Rather than viewing tissues as homogeneous units, single-cell approaches redefine them as structured communities of diverse and functionally specialized cells.
This paradigm shift has reshaped oncology, where tumor heterogeneity and immune microenvironment dynamics dictate treatment outcomes. This has extended into autoimmune disease, neurodegeneration, metabolic disorders, and developmental biology. The integration of spatial transcriptomics and multimodal profiling further enhances this resolution, linking transcriptional identity to physical context and regulatory state. Together, these advances are transforming disease classification from tissue-based frameworks to high-dimensional cellular atlases.
As the field progresses from discovery science toward clinical implementation, key challenges in standardization, interpretation, and regulatory validation must be addressed. Yet the trajectory is clear: high-resolution cellular mapping is redefining both biological inquiry and precision medicine.
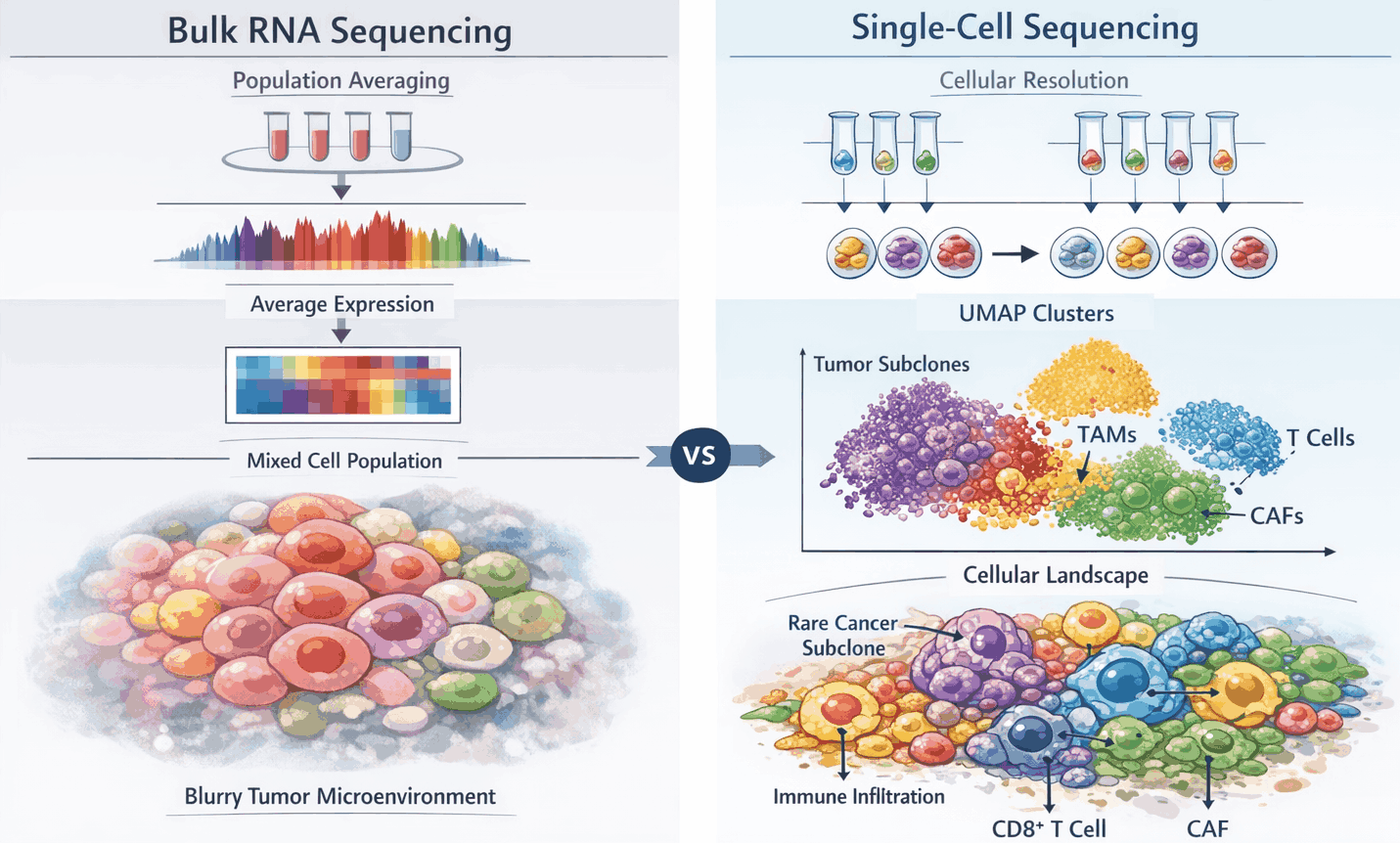
Figure 1: From bulk RNA sequencing to single-cell resolution in tumor ecosystems
Single-cell RNA sequencing (scRNA-seq) resolves the complexity of tumor ecosystems that bulk RNA sequencing cannot. Bulk RNA-seq averages gene expression across heterogeneous populations, obscuring rare malignant, immune, and stromal subpopulations. In contrast, scRNA-seq assigns unique molecular barcodes to each cell, enabling high-resolution profiling of individual transcriptomes. This approach allows identification of transcriptionally distinct malignant subclones, diverse immune subsets, and stromal cell states, providing mechanistic insights into tumor evolution, microenvironment interactions, and therapy resistance. Panels illustrate (A) bulk RNA-seq averaging heterogeneous cell populations, (B) scRNA-seq resolving distinct tumor, immune, and stromal compartments, and (C) reconstruction of cellular hierarchies and lineage trajectories.
Methods
Sample Preparation and Tissue Dissociation
Human tissue specimens are enzymatically and mechanically dissociated to obtain single-cell suspensions. For solid tissues, fragments are typically digested with collagenase (Types I–V) and trypsin, with incubation times ranging from 15 minutes at 37°C to overnight at room temperature, depending on tissue density. Protocols are optimized to minimize artificial transcriptional stress responses, as protease-based dissociation at elevated temperatures can induce stress-related gene expression. Blood-based samples, such as peripheral blood mononuclear cells (PBMCs), are thawed, washed in PBS, and resuspended in PBS containing 0.04% BSA. Target cell viability is generally >70%, and suspensions are filtered through 40 μm strainers to remove aggregates.
Single-Cell Capture and Barcoding
Single-cell capture can be achieved using various commercial strategies: droplet-based microfluidics, microwell arrays, and combinatorial indexing. In all approaches, unique molecular identifiers (UMIs) are appended to transcripts to distinguish original RNA molecules from PCR duplicates1.
Droplet-based systems encapsulate individual cells in hydrogel microspheres or oil droplets with barcoded beads. Microwell-based systems rely on gravity-driven deposition of single cells into micropores alongside barcoded beads. Combinatorial indexing employs split-pool barcoding on fixed and permeabilized cells, enabling high-throughput labeling without the use of microfluidic instruments.
Based on platform specifications, cell loading is adjusted to achieve target recovery rates.
Library Preparation and Amplification
Barcoded transcripts are reverse-transcribed into cDNA using reverse transcriptase with terminal transferase activity and a template-switch primer. Libraries are amplified via exponential PCR or linear in vitro transcription (IVT) to maintain transcript proportion. Random or template-switch primers are used depending on the platform. Final libraries are assessed for quality using standard tools such as Bioanalyzer, TapeStation, or Qubit fluorometry.
Sequencing
Libraries are sequenced using high-throughput platforms (e.g., Illumina NextSeq or NovaSeq) to a depth sufficient for downstream analysis, typically in the range of tens of thousands of reads per cell. Metrics such as read depth, mapping efficiency, and duplication rates are routinely monitored.
Computational Analysis
Raw reads are demultiplexed and processed with platform-specific pipelines. Quality control typically excludes cells with low gene detection or high mitochondrial content, and genes detected in very few cells are filtered out2. Normalization is performed using methods such as SCTransform or log-normalization. Dimensionality reduction is often performed using Principal Component Analysis (PCA), with batch effects corrected via tools such as Harmony3. Data is typically visualized using principal component analysis (UMAP), with graph-based clustering algorithms such as the Leiden4 community detection algorithm applied to delineate cellular subpopulations. Cell-type annotation can integrate automated approaches (e.g., CellTypist, Seurat5 label transfer) with manual inspection of marker genes.
Platform Comparison of Commercial scRNA-seq Technologies
Droplet-Based Microfluidics
Droplet-based platforms, such as 10x Genomics Chromium6 (3’, 5’, Fixed RNA Profiling) and Fluent PIPseq T20 v4.0, encapsulate individual cells with barcoded beads in hydrogel microspheres (GEMs). Each transcript is labeled with a unique molecular identifier (UMI), enabling distinction between original RNA molecules and PCR duplicates. The Fixed RNA Profiling (FRP) workflow additionally employs an oligonucleotide ligation assay (OLA) to detect transcripts in formaldehyde-fixed samples. These systems maximize molecular sensitivity, making them well-suited for detecting low-abundance transcripts in small populations. Target cell recovery per run typically ranges from 16,000 to 80,000 cells, depending on platform configuration.
Microwell-Based Arrays
Microwell-based technologies, including BD Rhapsody WTA, Honeycomb HIVE CLX, and Singleron GEXSCOPE, capture cells via gravity-driven deposition into microfabricated wells with barcoded beads. UMIs allow transcript tracking, while well-specific capture minimizes cross-contamination. These platforms provide a balance between throughput and sensitivity, offering recovery of 5,000–50,000 cells per run. Protocols vary in cDNA synthesis strategy: BD Rhapsody and Honeycomb use random priming, whereas Singleron incorporates template-switching mechanisms to generate full-length cDNA.
Combinatorial Indexing
Combinatorial indexing platforms, such as Parse Evercode WT v2 and Scale Single Cell RNA, bypass microfluidic partitioning by using split-pool barcoding to fix and permeabilize cells. Multiple rounds of ligation assign unique barcode combinations to each transcript. These methods achieve unmatched throughput, recovering hundreds of thousands of cells per experiment, though at slightly reduced molecular sensitivity compared with droplet-based workflows. Combinatorial indexing is particularly advantageous for large-scale atlasing projects or rare-cell enrichment across diverse tissue types.
Comparative Insights
Commercial single-cell RNA sequencing platforms vary in capture strategy, molecular sensitivity, throughput, and compatibility with fixed samples. Droplet- and microwell-based systems prioritize high transcript detection per cell, whereas combinatorial indexing platforms provide exceptional throughput suitable for large-scale mapping. Fixed-sample protocols (e.g., 10x Fixed RNA Profiling, Parse Evercode) enable retrospective studies, though often at the cost of reduced sensitivity.
Integration of these technologies with spatial and multi-omic modalities further shapes experimental design choices. Researchers must balance desired cell numbers, molecular resolution, and experimental constraints to select the most appropriate platform. The following tables, adapted from standardized benchmarking of human PBMCs by De Simone et al7, provide a quantitative framework for these decisions.
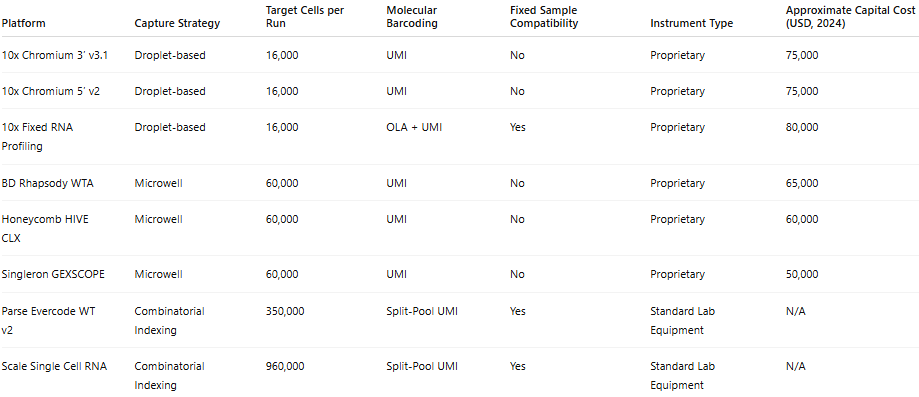
Table 1: Technical Characteristics of Commercial Single-Cell RNA Sequencing Platforms
Platforms are categorized according to capture strategy, including droplet-based microfluidics, microwell-based systems, and combinatorial indexing approaches. Technical features summarized include cell partitioning method, barcoding chemistry, transcript capture strategy (3′, 5′ or full-length), compatibility with formalin-fixed paraffin-embedded (FFPE) material, and relative throughput capacity. Information reflects manufacturer specifications and published methodological descriptions under standard operating conditions. These characteristics highlight fundamental architectural differences that influence scalability, sensitivity, and experimental flexibility across platforms.
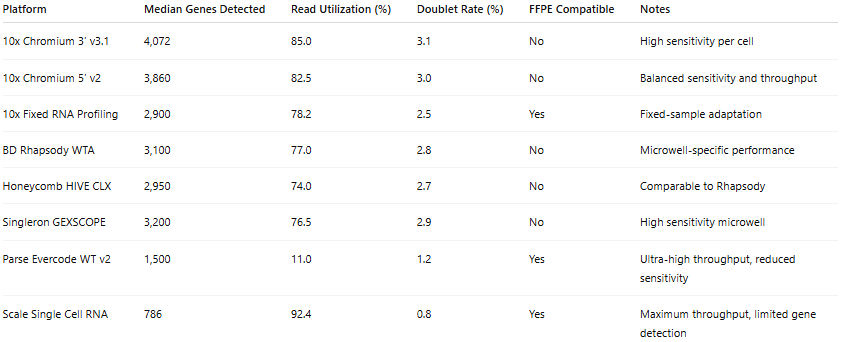
Table 2: Benchmark Performance Under Standardized PBMC Conditions
Platforms were evaluated using human peripheral blood mononuclear cells processed under harmonized experimental and computational workflows. Median genes detected per cell, read utilization efficiency, and estimated doublet rates were derived from standardized benchmarking analyses reported by De Simone et al. Sequencing depth ranged from approximately 30,000 to 50,000 reads per cell. FFPE compatibility indicates whether formalin-fixed paraffin-embedded material is compatible with the platform chemistry. Doublet rates were estimated using simulation-based approaches under controlled cell-loading conditions. These data provide a comparative framework for assessing sensitivity, efficiency, and sample compatibility across droplet-based, microwell-based, and combinatorial indexing systems.
Oncology Applications of Single-Cell RNA Sequencing
Oncology has served as the clearest demonstration of the single-cell technological inflection. Tumors, once understood primarily as genetically heterogeneous masses, are now recognized as structured cellular ecosystems in which malignant, immune, stromal, and vascular compartments dynamically interact. Single-cell transcriptomics has shifted cancer biology from clonal population models to state-resolved ecosystem mapping, fundamentally altering how tumor progression, immune evasion, and therapeutic resistance are conceptualized.
Single-cell RNA sequencing (scRNA-seq) enables high-resolution characterization of cellular heterogeneity within the tumor microenvironment by assigning molecular barcodes to individual transcriptomes, allowing thousands of cells to be profiled simultaneously while preserving cell-of-origin identity. Unlike bulk approaches that average gene expression across mixed populations, scRNA-seq resolves malignant and non-malignant compartments at cellular resolution, facilitating the identification of rare subclones, transitional states, and therapy-resistant populations that would otherwise remain obscured.
Despite its transformative resolution, single-cell transcriptomics remains susceptible to dissociation-induced transcriptional artifacts and gene dropout, which can distort inferred cellular hierarchies. Careful experimental design and orthogonal validation are therefore essential to distinguish biologically meaningful heterogeneity from technical noise.
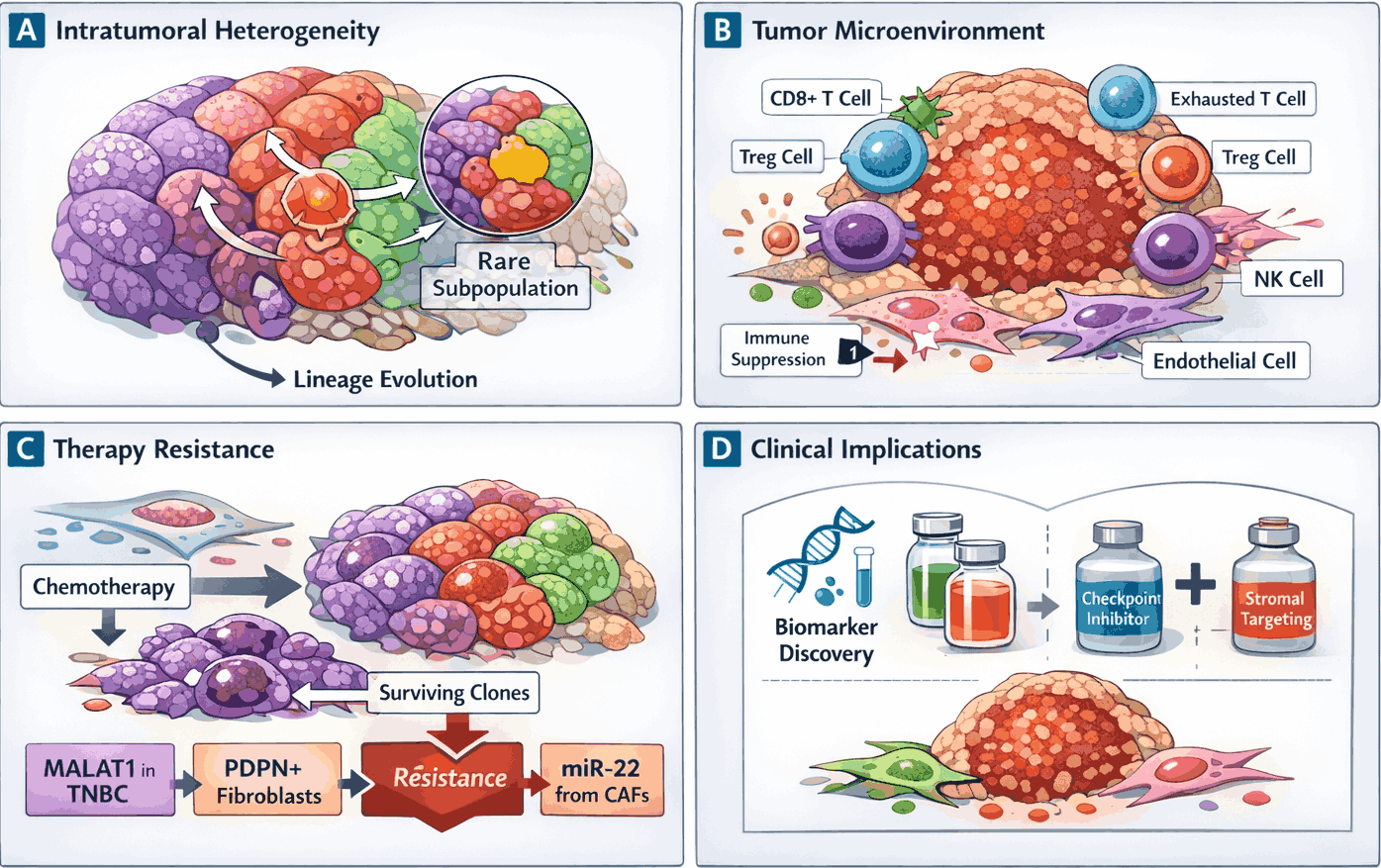
Figure 2: Single-cell RNA sequencing reveals tumor heterogeneity and microenvironment complexity
Single-cell RNA sequencing (scRNA-seq) dissects the cellular composition and transcriptional states within tumors. (A) Malignant subpopulations exhibit transcriptionally distinct states and lineage trajectories, revealing rare and aggressive clones. (B) The tumor microenvironment comprises immune (CD8⁺ T, regulatory T, natural killer (NK) cells) and stromal (cancer-associated fibroblasts, endothelial) compartments, with immune suppression and T cell exhaustion indicated. (C) Transcriptional heterogeneity underlies therapy resistance, with resistant subpopulations driven by long non-coding RNAs (MALAT1), podoplanin-positive CAFs, or cancer-associated fibroblast (CAF)-derived miRNAs. (D) High-resolution single-cell maps guide biomarker discovery and inform rational combination therapy strategies.
Breast Cancer
scRNA-seq reveals extensive cellular diversity within breast tumors, uncovering transcriptionally distinct malignant subpopulations and transitional states that drive progression and therapeutic resistance. Aggressive clones often exhibit a luminal progenitor (LP) signature, associated with high copy-number alterations and serving as a potential cell of origin for multiple breast cancer types8. Signaling pathways driving these populations include HER2, PI3K/AKT/mTOR, and estrogen receptor pathways. The SCSubtype classification identifies diverse ecotypes across Luminal, HER2+, and TNBC tumors, with Met-E subtypes exhibiting the highest intratumor heterogeneity and poorest prognosis9.
The tumor microenvironment varies by subtype. TNBC, for example, features immunosuppressive niches with reduced CD8⁺ T cells and expanded regulatory T cells, driven by PD-L1 and TGF-β. Stromal CAFs, including ECM-myCAFs (DPP4/YAP-1 dependent), promote immunosuppression by polarizing TREM2⁺ macrophages and regulatory NK cells10. Exhausted T cells show high PDCD1 expression and elevated dysfunction scores.
Therapy resistance arises from transcriptional heterogeneity. MALAT1 drives resistance to neoadjuvant chemotherapy in triple-negative breast cancer (TNBC)11, while CD63⁺ CAFs in luminal tumors confer tamoxifen resistance via exosomal miR-22, which inhibits PTEN and activates PI3K/AKT112. Podoplanin-positive CAFs reduce trastuzumab efficacy in HER2+ tumors by secreting immunosuppressive factors that inhibit NK cell–mediated cytotoxicity13. Single-cell analyses identify gene programs associated with resistance and support rational combination design therapy, such as combining STING agonists with anti-TGF-β/PD-L1 antibodies or targeting stromal components in aggressive TNBC. Mast cell GATA3 expression predicts high residual cancer burden after neoadjuvant therapy, enabling subtype-specific intervention14.
Ovarian Cancer
scRNA-seq reveals transcriptionally distinct malignant subpopulations and transitional states in ovarian tumors, informing tumor evolution and plasticity. Markers such as MUC16 (CA125) identify malignant populations, while SOX9⁺LGR5⁺ signatures mark aggressive clones associated with high copy-number alterations15.
Profiling the tumor microenvironment delineates diverse immune and stromal populations, including CAFs, endothelial cells, and myeloid subsets. Immunosuppressive niches are dominated by M2-type tumor-associated macrophages (TAMs) in autophagic states, releasing oncostatin M (OSM) under IL-6 influence to activate STAT3 signaling in tumor cells, driving invasion and metastasis16. T cell exhaustion is promoted by IL-15 and CD47, leading to the accumulation of CD8⁺ terminally exhausted cells expressing CTLA4, PDCD1, and LAG317.
Transcriptional heterogeneity underlies therapy resistance. Resistant populations pre-exist or emerge adaptively. Cisplatin resistance is driven by SLAMF7 and GNAS expression in TAMs and by circular RNA ITGB6, which reprograms macrophages toward pro-tumor phenotypes18. CAFs contribute via TGF-β–mediated secretion of IL-6, promoting neovascularization, and RGS5⁺ CAFs form physical barriers that limit cytotoxic immune cell infiltration19. scRNA-seq identifies these gene programs, informing combination therapies targeting malignant and microenvironmental contributors to resistance.
Endometrial Cancer
scRNA-seq reveals that endometrial cancer exhibits a less differentiated epithelial phenotype, with malignant populations defined by SOX9⁺ LGR5⁺ and SOX9⁺ LGR5⁻ signatures20. SOX9⁺ LGR5⁺ clones predominate in aggressive serous and high-grade endometrioid subtypes, correlating with the Cancer Genome Atlas (TCGA) CNV-high categories and advanced stages. These clones are driven by dysregulated PI3K signaling, DNA mismatch repair, and Wnt/β-catenin pathways, particularly in p53-abnormal tumors. Tumorigenesis also involves LCN2⁺ and SAA1/2⁺ cells from unciliated glandular epithelium and clusters with heightened estrogen signaling via MUC1 and ELF3.
The microenvironment includes CAFs and immune cells. Four CAF subclusters, inflammatory, antigen-presenting, matrix, and vascular, support malignant stemness via ALDH1, AXIN2, and MYC21, with MDK–NCL signaling mediating epithelial–stromal crosstalk22. Immune components include CD8⁺ tissue-resident memory T cells, NK1–3 subsets (with NK2 showing intermediate maturation), and pro-tumorigenic CXCL8^hiIL1B^hi macrophages secreting SPP1 and NAMPT.
Therapy resistance is linked to transcriptional heterogeneity, epithelial–mesenchymal transition (EMT), and hypoxia signaling. Distal regulatory elements at IMPA2 and SOX9 loci correlate with elevated malignant expression. MMRd tumors show subtype-specific responses: mutational mismatch repair–deficient (MMRd) via CD8⁺ effectors, epigenetic MMRd via CD16⁺ NK cells23. Tertiary lymphoid structures (TLS) marked by L1CAM indicate robust anti-tumor immunity, particularly in POLE-mutated and MMRd tumors24.
Translational insights include biomarkers HSPA1B, TFF3, LAMA4, and a macrophage signature of SLC8A1, TXN, and CD74 for patient stratification. Targeting Netrin-1 to inhibit EMT25 or disrupting MDK–NCL signaling addresses stromal-mediated resistance. In CNV-high subsets, CDK12 and SMARCA4 are actionable targets for approved therapies26, enabling personalized interventions based on specific endometrial tertiary tumor microenvironment (TME) ecotypes.
Beyond Cancer
Single-cell RNA sequencing (scRNA-seq) has transformed the study of diverse biological systems by resolving cellular heterogeneity and defining rare and transitional cell states. By profiling thousands to millions of cells, scRNA-seq enables the reconstruction of lineage hierarchies, identification of developmental trajectories, and characterization of context-dependent transcriptional programs. These insights provide mechanistic understanding and translational opportunities across immunology, developmental biology, cardiovascular, neurological, and metabolic systems.
Immune System Profiling
The application of scRNA-seq has revolutionized immunology by resolving functional states of heterogeneous immune populations previously obscured in bulk analyses. The technology identifies fine-grained subsets, such as NK cells categorized into NK1 and NK2 subclasses, and reveals novel dendritic cell types capable of unique T lymphocyte activation. In chronic infections and autoimmune conditions, scRNA-seq uncovers activation and exhaustion states, exemplified by reduced cytotoxicity of CD8⁺ T cells in IgA nephropathy27 or the expansion of follicular helper T (Tfh) cells that support B-cell responses via CD40–CD40L signaling28. Moreover, not all transcriptionally defined cell states are functionally or clinically actionable. Integrating transcriptomic observations with spatial, proteomic, and longitudinal validation is critical to distinguish molecular heterogeneity that truly drives disease from transient or context-specific states. These high-resolution immune maps have profound translational relevance, informing vaccine development and personalized immunotherapy by enabling the identification of latent versus active cellular programs and predicting patient-specific therapeutic responses.
Development and Regeneration
scRNA-seq enables high-resolution mapping of lineage hierarchies and developmental trajectories. In embryogenesis, it reveals epigenetic and transcriptomic processes governing early mammalian development. While in adult tissues, it captures sequential cell fate transitions during human spermatogenesis or retinal organoid maturation. Physical context and intercellular interactions are critical; for example, fibro-adipogenic progenitors and macrophages cooperate in skeletal muscle regenerative niches. Translational applications include identifying candidate targets for cell-based therapies and lineage tracing using tools such as LinTIMaT29, which links transcriptomic data with mutational lineage, enabling early intervention in developmental disorders. Chromatin potential analysis further predicts future gene expression states, providing mechanistic insight for regenerative strategies30.
Neurological and Cardiac Applications
In the nervous system, scRNA-seq resolves neuronal and glial diversity and identifies disease-specific states. Parkinson’s disease is associated with CADPS2-overexpressing neuronal clusters31, and multiple sclerosis exhibits at least ten microglial subtypes emerging during disease progression32. Cardiac scRNA-seq characterizes fibroblast and vascular smooth muscle heterogeneity in heart failure and atherosclerosis, uncovering pathogenic populations driving remodeling. Spatial and interaction-aware techniques, such as RABID-seq33, reveal key signaling pathways such as Sema4D–PLXNB1 mediating microglia–astrocytic crosstalk. These high-resolution maps inform stem cell therapies, biomarker discovery, and regenerative interventions, including the use of induced pluripotent stem cells (iPSC) derived dopaminergic neurons or angiogenic endothelial populations in Alzheimer’s disease.
Inflammatory and Metabolic Disorders
scRNA-seq dissects cell-type-specific transcriptional programs driving chronic inflammatory and metabolic diseases. In metabolic disorders, DPP4⁺ adipocyte progenitors regulate differentiation and insulin sensitivity34, whereas in IgA nephropathy, mesangial heterogeneity predicts disease progression. The microenvironment involves intricate crosstalk; in rheumatoid arthritis, fibroblast subsets expressing CXCL12 and matrix metalloproteinases drive joint destruction, while in Metabolic dysfunction–associated steatohepatitis (MASH), resident Kupffer cells and infiltrating macrophages exert distinct pathogenic roles. Translationally, these insights enable personalized strategies: the GIMATS cellular module predicts anti-TNF therapy resistance in Crohn’s disease35, and kidney podocyte signatures, including CD74 and B2M, identify potential therapeutic targets to prevent renal failure.
Across these systems, scRNA-seq provides mechanistic insight into cellular hierarchies, microenvironmental interactions, and disease-specific transcriptional programs, extending the precision and translational potential of single-cell approaches far beyond oncology.
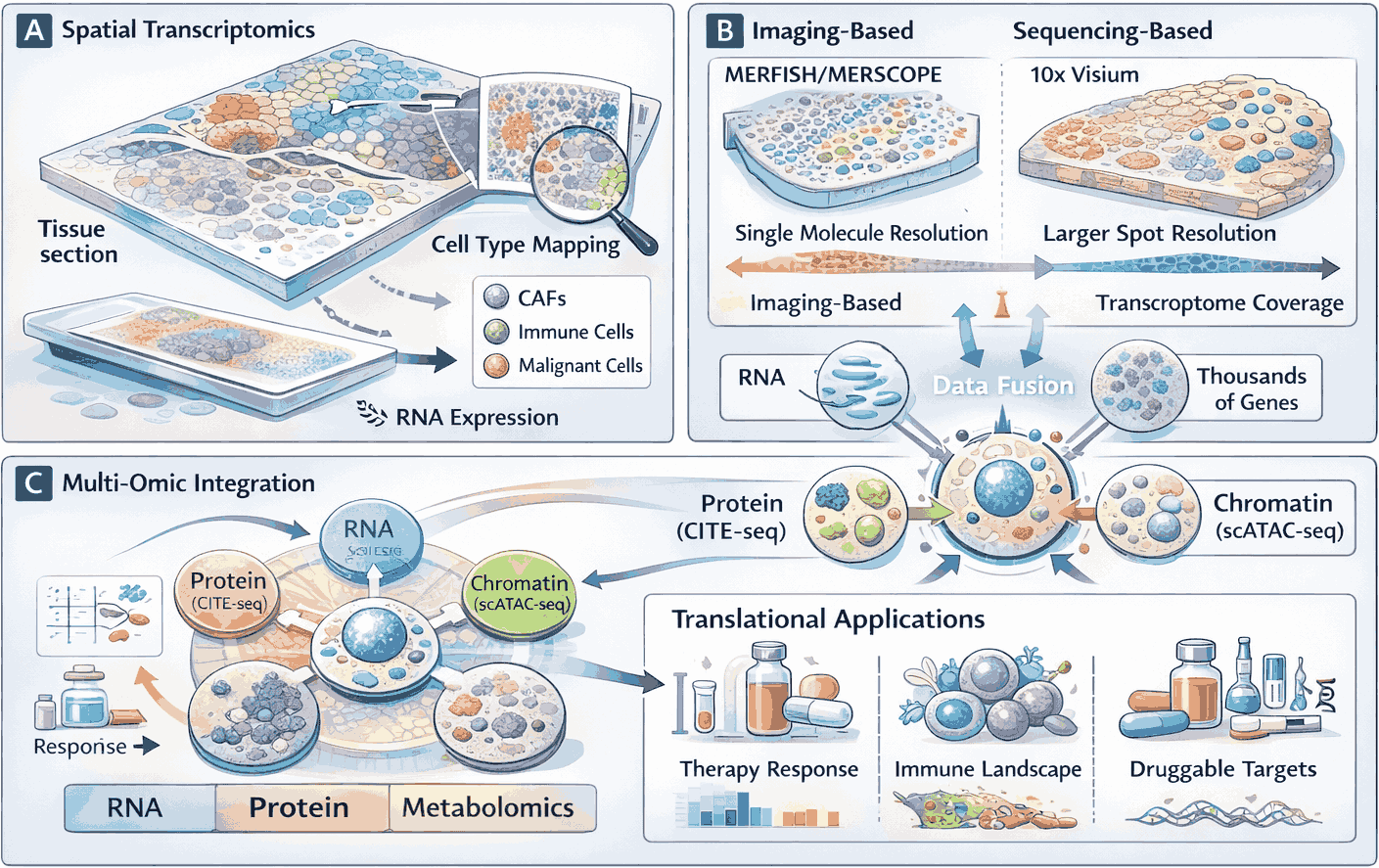
Figure 3: Spatial and Multi-Omic Integration of Single-Cell Data to Inform Translational Medicine
Single-cell transcriptomics and multi-omic profiling are increasingly integrated with spatial resolution to preserve tissue context while capturing molecular complexity. (A) Spatial transcriptomics resolves distinct cell populations within their native microenvironment. (B) Comparison of imaging-based and sequencing-based spatial approaches highlighting resolution and transcriptome coverage trade-offs. Middle: Multi-omic integration captures RNA, protein, chromatin, and metabolic states within the same cells, enabling comprehensive molecular characterization. (C) Translational applications, including prediction of therapy response and identification of druggable targets, illustrate the clinical potential of spatial and multi-omic single-cell approaches.
Future Directions: Spatial Architecture and Multimodal Integration
Although scRNA-seq provides high-resolution transcriptomic profiles, tissue dissociation disrupts spatial architecture and intercellular context. Integration of spatial and multi-omic approaches is transforming single-cell research through modalities including high-plex RNA imaging (for example, MERSCOPE/MERFISH and seqFISH⁺), spatial barcoding (for example, 10x Visium and Slide-seq), and multimodal single-cell technologies such as CITE-seq (RNA and surface proteins) and scATAC-seq (chromatin accessibility). Biological function is fundamentally dependent on a cell’s physical location and its interactions with neighboring populations.
Despite these technological advances, the majority of single-cell studies still rely on dissociated tissues, which may obscure transient or context-specific cell states. Validating transcriptomic observations through orthogonal modalities, such as protein measurements or spatial mapping, is critical to distinguish functional cellular programs from noise or technical artifacts. Integrating multi-omic and spatial information allows researchers to define clinically actionable cell states, linking transcriptional identity to location, signaling context, and therapeutic relevance. This multi-layered validation ensures that observed heterogeneity reflects true biological variation rather than experimental or dissociation-induced artifacts.
Spatial Transcriptomics: Preserving the Tissue Landscape
Spatial transcriptomics identifies RNA molecules within their native tissue context, providing insight into how microenvironmental positioning shapes cellular function. Imaging-based platforms, such as MERFISH and seqFISH⁺, achieve subcellular resolution through multiplexed fluorescent probe hybridization to predefined RNA targets36. Sequencing-based approaches, including 10x Visium37, use spatially barcoded capture arrays to profile transcriptomes across tissue sections; however, each capture spot may encompass multiple cells.
Spatial mapping has revealed functionally relevant tissue organization. In breast cancer, distinct CAF spatial zones correlate with prognosis38, and the localization of CD8⁺ tissue-resident memory T cells predicts immunotherapy response. In ovarian cancer, CAFs form physical barriers that impede cytotoxic immune infiltration in short-survival patients39. Probe-based approaches now enable studies in archival FFPE specimens, facilitating high-resolution retrospective analyses of rare or aggressive disease subtypes.
Multi-Omics Integration: Resolving Complex Molecular States
No single modality captures the full complexity of cellular identity. Multi-omic approaches integrate transcriptomics with proteomics, epigenomics, and metabolomics to resolve regulatory programs and predict future cell states. CITE-seq measures RNA and surface proteins simultaneously40, while scATAC-seq and 10x Multiome profile chromatin accessibility alongside transcriptomes. These integrations reveal phenomena invisible to transcriptomics alone, such as protein-RNA discordance41 or epigenetic priming of gene expression.
Mechanistic insights include chromatin accessibility at IMPA2 and SOX9 loci in endometrial cancer, predicting malignant gene expression and therapy resistance42. Single-cell metabolomics can guide drug repurposing, as exemplified by mifepristone in melanoma. Clinically, deep-learning frameworks integrating multi-omic immune cell profiles predict drug responses and patient survival in breast cancer. Combining scTCR-seq with scRNA-seq further links immune clonotypes to transcriptional states, refining CAR-T therapy strategies.
Challenges and Outlook
Despite rapid technological advances, the routine clinical implementation of single-cell and spatial multi-omic platforms remains limited. Multi-omic and spatial platforms are constrained by limited throughput, high cost, and temporal asynchrony between molecular layers. The resulting high-dimensional datasets necessitate advanced computational frameworks and standardized analytical pipelines to translate complex cellular atlases into clinically actionable insights. Future efforts will prioritize methodological standardization, enhanced spatial resolution and molecular coverage, and integration of multi-omic readouts into robust predictive models for precision medicine. Achieving clinical scalability will require harmonized protocols, cross-platform validation, and regulatory frameworks capable of accommodating high-dimensional molecular diagnostics.
Clinical Translation: From Bench to Bedside
The transition of single-cell RNA sequencing (scRNA-seq) from a discovery-oriented research tool to a clinical diagnostic platform represents a critical inflection point in modern biology. Originally developed to resolve cellular heterogeneity in research settings, scRNA-seq now demands a shift in priorities from cellular breadth to analytical robustness. Whereas discovery-focused applications emphasize identification of novel cell states, clinical diagnostics require reproducibility, rapid turnaround times, standardized workflows, and regulatory validation.
Several fundamental barriers, including high per-sample costs, lack of standardized protocols, and the complexity of data interpretation, must be overcome before single-cell profiling can become routine in medical care.
Technical and Operational Barriers
Current high-resolution scRNA-seq assays typically cost US $500–1,000 per sample, exceeding reimbursement thresholds for most established molecular diagnostic tests. Sequencing efficiency remains variable, with some platforms utilizing less than 40% of generated reads, thereby increasing the effective sequencing depth required per sample. Clinical timelines impose additional constraints: diagnostic decisions often require results within 24–72 hours, whereas current scRNA-seq workflows may require 5–14 days from tissue acquisition to analysis. Sample integrity further complicates implementation: droplet-based methods generally require fresh tissue, and cold chain logistics are critical to prevent artifactual transcriptional stress responses or cell death. The development of targeted gene panels and probe-based hybridization methods, such as 10x Fixed Genomics, enables the use of archival FFPE specimens, partially mitigating these logistical challenges.
Regulatory and Standardization
Regulatory and standardization hurdles remain substantial. Most clinical single-cell applications are performed as Laboratory Developed Tests (LDTs) rather than kits cleared by the U.S. Food and Drug Administration (FDA), reflecting the evolving nature of the field. Batch effects, variable tissue processing workflows, and inconsistencies in quality-control thresholds across laboratories influence reproducibility. International consortia, such as the Human Cell Atlas43, are establishing reference atlases of healthy tissues to define baseline cellular states and improve cross-study comparability. The development of universal quality-control metrics is essential to ensure analytical consistency across clinical sites and multicenter trials. Including minimum gene detection and maximum mitochondrial read fractions is critical to ensure comparability across clinical sites and trials.
Data Analysis Complexity and Patient Stratification
Despite these challenges, scRNA-seq is already informing patient stratification and early translational studies. In Crohn’s disease, single-cell analysis identified the GIMATS cellular module, which predicts resistance to anti–tumor necrosis factor (anti-TNF) therapy, while in Type 1 diabetes, the T1DM metagene z-score allows molecular subtyping that correlates with disease risk and treatment response. Demonstrating the convergence of high-dimensional biology and computational oncology. Hematologic malignancies, particularly acute myeloid leukemia, due to the relative ease of sampling and the established diagnostic utility of immune cell subtyping, scRNA-seq can detect minimal residual disease by identifying rare relapsing clones that evade flow cytometry44.
Early Clinical Implementations and Trials
Several oncology trials illustrate the emerging clinical utility of scRNA-seq. The BMT CTN 1401 Phase II trial in multiple myeloma uses single-cell profiling to track the expansion of myeloma-reactive CD8⁺ T cells following dendritic cell/myeloma fusion vaccination combined with lenalidomide maintenance. In operable non-small cell lung cancer, the NEOSTAR Phase II trial45 leverages scRNA-seq to evaluate the immunological impact of neoadjuvant chemotherapy combined with immune-checkpoint blockade46 on the behavior of CD8⁺ T, CD4⁺ T, B, and NK cells within the tumor. Similarly, in a colon cancer trial evaluating combined PD-1, BRAF, and MEK inhibition, single-cell analysis revealed enhanced interferon signaling and T cell recruitment, providing mechanistic insight into therapeutic efficacy. In solid tumors such as endometrial cancer, scRNA-seq has identified CNV-high subtypes with gene products such as CDK12 and SMARCA4 as actionable targets for existing FDA-approved drugs.
Outlook
While significant technical, regulatory, and operational barriers remain, the per-cell cost of sequencing is declining, and analytical pipelines are maturing. The integration of high-resolution single-cell maps into clinical practice is advancing, particularly in oncology and immunology, where cellular heterogeneity directly influences therapeutic response. These advances promise to guide targeted therapy development, improve patient stratification, and ensure that treatment strategies are aligned with the specific cellular drivers of disease.
Conclusion
Single-cell RNA sequencing has redefined the resolution at which biology and disease are understood. By moving beyond population-averaged measurements to cell-state resolved profiling, this technology has transformed tissues from homogeneous entities into dynamic ecosystems composed of interacting and functionally specialized cellular compartments. What began as a methodological advance has evolved into a conceptual shift in how pathology is framed, classified, and interrogated.
In oncology, single-cell approaches have deconstructed tumor heterogeneity, revealed mechanisms of immune evasion and therapeutic resistance, and identified rare subclones that drive relapse. Beyond cancer, these technologies have uncovered pathogenic cell states in autoimmune disease, clarified cellular vulnerability in neurodegeneration, and resolved developmental trajectories with unprecedented precision. The integration of spatial transcriptomics and multimodal profiling has further expanded this framework, linking transcriptional identity to physical context, epigenetic regulation, and protein expression. Collectively, these advances mark a transition from descriptive cellular cataloging to systems-level modeling of cellular ecosystems.
The clinical translation of single-cell genomics represents the maturation of this technological inflection. Although economic, computational, and regulatory barriers remain, early applications in oncology and immunology demonstrate the feasibility of incorporating high-dimensional cellular maps into therapeutic decision-making. As analytical pipelines become standardized and costs decline, the distinction between exploratory single-cell research and clinical molecular diagnostics is likely to be too narrow.
Ultimately, the promise of single-cell genomics lies not simply in identifying new cell types but in redefining disease classification itself. By resolving pathology at cellular resolution, these approaches challenge traditional organ-based and histological frameworks, advancing medicine toward ecosystem-aware, mechanism-driven stratification. As spatial, multimodal, and computational innovations continue to converge, the capacity to map, predict, and therapeutically modulate cellular systems will expand, positioning high-resolution cellular atlases as foundational components of precision medicine in the decade ahead.
References
1. Andersson, D., Kebede, F. T., Escobar, M., Österlund, T. & Ståhlberg, A. Principles of digital sequencing using unique molecular identifiers. Mol. Aspects Med. 96, 101253 (2024).
2. Luecken, M. D. & Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746 (2019).
3. Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
4. Va, T., L, W. & Nj, van E. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, (2019).
5. Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573-3587.e29 (2021).
6. Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
7. Simone, M. D. et al. Comparative Analysis of Commercial Single-Cell RNA Sequencing Technologies. 2024.06.18.599579 Preprint at https://doi.org/10.1101/2024.06.18.599579 (2024).
8. Pal, B. et al. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. 40, e107333 (2021).
9. Wu, S. Z. et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 53, 1334–1347 (2021).
10. Bartoschek, M. et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 9, 5150 (2018).
11. Kim, C. et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 173, 879-893.e13 (2018).
12. Hu, J. et al. Single-Cell Transcriptome Analysis Reveals Intratumoral Heterogeneity in ccRCC, which Results in Different Clinical Outcomes. Mol. Ther. J. Am. Soc. Gene Ther. 28, 1658–1672 (2020).
13. Kieffer, Y. et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 10, 1330–1351 (2020).
14. Yeo, S. K. et al. Single-cell RNA-sequencing reveals distinct patterns of cell state heterogeneity in mouse models of breast cancer. eLife 9, e58810 (2020).
15. Izar, B. et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 26, 1271–1279 (2020).
16. Bi, K. et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 39, 649-661.e5 (2021).
17. Jiménez-Sánchez, A. et al. Unraveling tumor-immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat. Genet. 52, 582–593 (2020).
18. Zhao, Q., Shao, H. & Zhang, T. Single-cell RNA sequencing in ovarian cancer: revealing new perspectives in the tumor microenvironment. Am. J. Transl. Res. 16, 3338–3354 (2024).
19. Hao, D. et al. Integrated Analysis Reveals Tubal- and Ovarian-Originated Serous Ovarian Cancer and Predicts Differential Therapeutic Responses. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 23, 7400–7411 (2017).
20. González-Martínez, S., Pérez-Mies, B., Cortés, J. & Palacios, J. Single-cell RNA sequencing in endometrial cancer: exploring the epithelial cells and the microenvironment landscape. Front. Immunol. 15, 1425212 (2024).
21. Deng, C.-C. et al. Single-cell RNA-seq reveals fibroblast heterogeneity and increased mesenchymal fibroblasts in human fibrotic skin diseases. Nat. Commun. 12, 3709 (2021).
22. Nair, H. B. et al. Midkine (MDK) as a central regulator of the tumor microenvironment: From developmental cytokine to therapeutic target. Cancer Lett. 641, 218258 (2026).
23. Chow, R. D. et al. Distinct mechanisms of mismatch repair deficiency delineate two modes of response to PD-1 immunotherapy in endometrial carcinoma. Cancer Discov. 13, 312–331 (2023).
24. Cabrita, R. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020).
25. Lengrand, J. et al. Pharmacological targeting netrin-1 inhibits EMT in cancer. Nature 620, 402–408 (2023).
26. Liu, W. et al. The landscape and prognostic value of immune characteristics in uterine corpus endometrial cancer. Biosci. Rep. 41, BSR20202321 (2021).
27. Xia, M., Li, Y., Liu, Y., Dong, Z. & Liu, H. Single-Cell RNA-Sequencing Analysis Provides Insights into IgA Nephropathy. Biomolecules 15, 191 (2025).
28. Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 50, 1132–1148 (2019).
29. Yang, D. et al. Lineage tracing reveals the phylodynamics, plasticity, and paths of tumor evolution. Cell 185, 1905-1923.e25 (2022).
30. Buenrostro, J. D. et al. Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 173, 1535-1548.e16 (2018).
31. Smajić, S. et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain J. Neurol. 145, 964–978 (2022).
32. Absinta, M. et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 597, 709–714 (2021).
33. Chan, M. M. et al. Molecular recording of mammalian embryogenesis. Nature 570, 77–82 (2019).
34. Emont, M. P. et al. A single-cell atlas of human and mouse white adipose tissue. Nature 603, 926–933 (2022).
35. Martin, J. C. et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 178, 1493-1508.e20 (2019).
36. Zhang, M. et al. Spatially resolved cell atlas of the mouse primary motor cortex by MERFISH. Nature 598, 137–143 (2021).
37. Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
38. Bassez, A. et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med. 27, 820–832 (2021).
39. Mhaidly, R. & Mechta-Grigoriou, F. Fibroblast heterogeneity in tumor micro-environment: Role in immunosuppression and new therapies. Semin. Immunol. 48, 101417 (2020).
40. Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017).
41. Liu, Y., Beyer, A. & Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165, 535–550 (2016).
42. Dou, J. et al. Bi-order multimodal integration of single-cell data. Genome Biol. 23, 112 (2022).
43. Regev, A. et al. The Human Cell Atlas. eLife 6, e27041 (2017).
44. Álvarez, N. et al. Detection of minimal residual disease in acute myeloid leukemia: evaluating utility and challenges. Front. Immunol. 15, 1252258 (2024).
45. Cascone, T. et al. Neoadjuvant nivolumab or nivolumab plus ipilimumab in operable non-small cell lung cancer: the phase 2 randomized NEOSTAR trial. Nat. Med. 27, 504–514 (2021).
46. Cascone, T. et al. Neoadjuvant nivolumab or nivolumab plus ipilimumab in operable non-small cell lung cancer: the phase 2 randomized NEOSTAR trial. Nat. Med. 27, 504–514 (2021).